
This article covers
What are lysosomes? A quick overview
The lysosomes are small organelles that work as the recycling center in the cells. They are membrane-bounded spheres full of digesting enzymes. These enzymes can break down whatever substance (usually, old cell parts) entering the lysosomes into small molecules (amino acids, nucleotides, fatty acids, and sugars), so the cell can reuse these raw materials to build new organelles. Lysosomes may also be used to destroy invading viruses and bacteria.
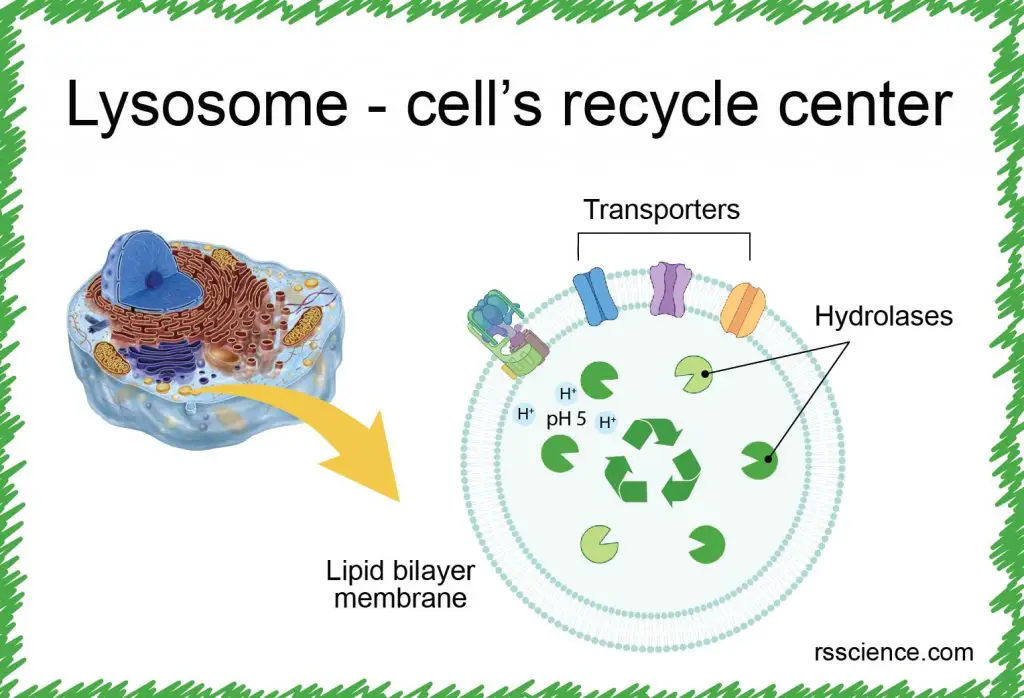
[In this figure] Lysosome analogy: lysosome is the recycling center of the cell.
It stores all kinds of enzymes capable of breaking down proteins, nucleic acids, carbohydrates, and lipids. You can also say that lysosomes are like the human stomach. The stomach is where food is digested and broken down for absorption.
What does lysosome look like?
Lysosomes are present in almost all eukaryotic cells except red blood cells. Lysosomes locate in the cytoplasm. They display considerable variation in size and shape. In regular cells, lysosomes are spherical bodies about 50-70 nm in diameter. Several hundred lysosomes may be present in a single animal cell. These lysosomes are too small to be seen under a regular light microscope. Electron microscope or fluorescence microscope are required to observe and study lysosomes.

[In this figure] Fluorescence microscopic image of lysosomes (red) and autophagosomes (green).
Photo source: AAT Bioquest.
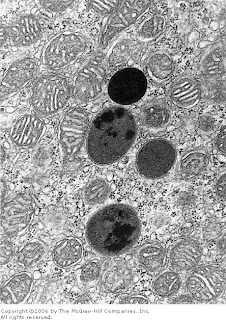
[In this figure] Electron micrograph (EM) showing four dark secondary lysosomes surrounded by numerous mitochondria.
Photo source: De Histology
Lysosomes are particularly abundant in cells exhibiting phagocytic activity (eg, macrophages, neutrophiles, and kidney cells). In these cells, their primary lysosomes are larger, up to 0.5 micrometer in diameter, and thus are just visible with the light microscope.
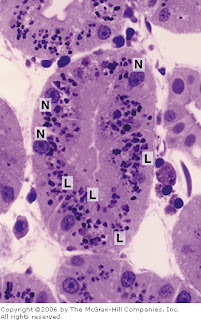
[In this figure] Picture of a toluidine blue-stained kidney tubule.
Large lysosomes (L) are abundant in these kidney cells. (N) is the cell nucleus.
Photo source: De Histology

[In this figure] Neutrophils are in charge of destroying bacteria by phagocytosis.
Image source: Cellcartoons
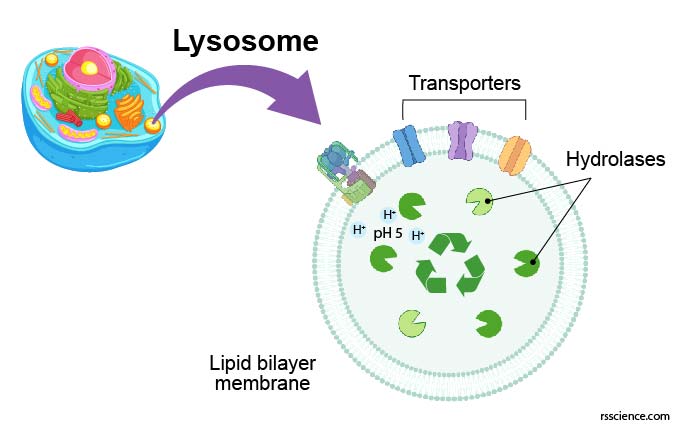
The structure of lysosome
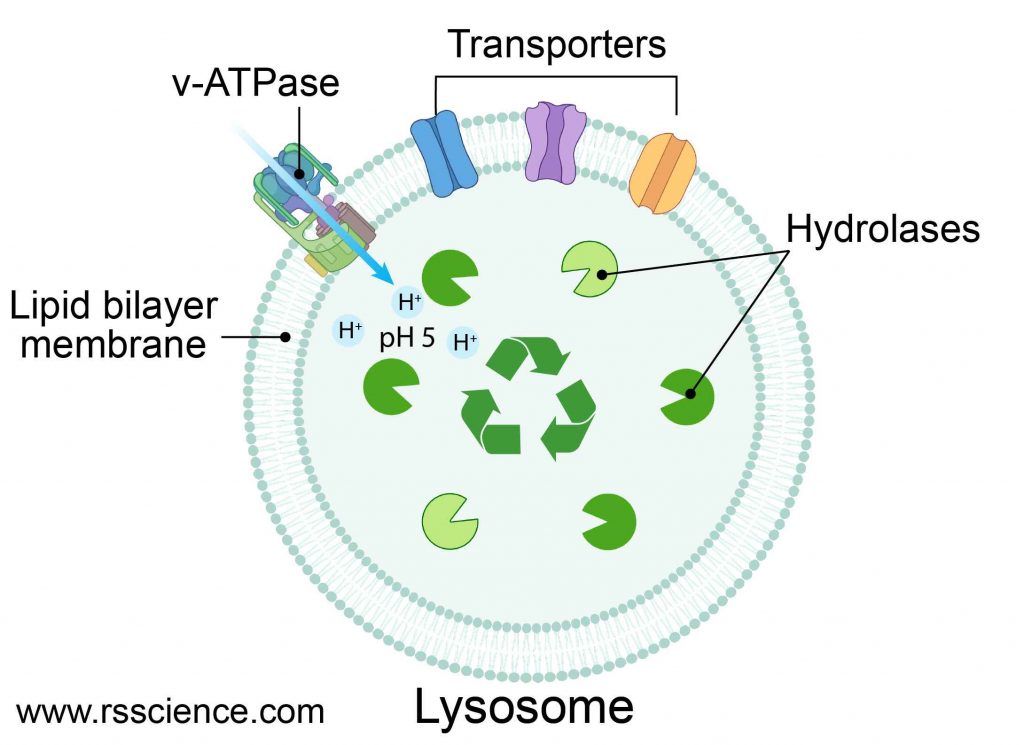
[In this figure] Structure diagram of lysosomes.
The lysosome is like a bag that consisted of a single layer of the lipid bilayer membrane. On the membrane, there are ion bumps that can maintain an acidic internal environment of the lysosome. Inside the bag, there are many digestive enzymes.
The lysosome is a membrane-bound vesicle containing hydrolase enzymes that break down old organelles and proteins into small molecules, such as amino acids. Lysosome’s membrane is similar to the cell membrane or endoplasmic reticulum (ER) since lysosomes are budded from the ER-Golgi system. The membrane surrounding the lysosome is vital to ensure these enzymes do not leak out into the cytoplasm and damage the cell from within.
Several proteins are embedded in the lysosome’s membrane, including v-ATPase that pumps protons into the lysosome to acidify its pH value. There are also several types of transporters that export recycled nutrients (e.g., sugars, nucleic acids, and amino acids) from lysosomes back to the cytoplasm.
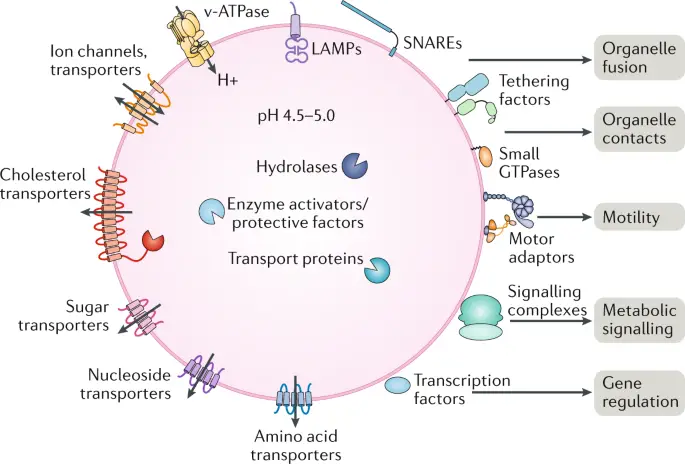
[In this figure] There are many transporter proteins across the membrane of lysosomes. They can pump H+ inward to maintain acidic environments for hydrolase enzymes. Corresponding transporters can also export recycled nutrients back to the cytoplasm.
Photo source: Nature reviews Molecular cell biology.
Lysosome enzymes and acidic environment
Lysosomes contain about 50 different degradative enzymes that can hydrolyze proteins, DNA, RNA, polysaccharides, and lipids. The product of lysosome digestion can be recycle back for cell to build new organelles.
As you can imagine, if the lysosomes somehow leak or burst, these enzymes could cause huge damages to the cells. Fortunately, the cells have a safety mechanism to restrict lysosomal enzymes to be active only in an acidic environment, but not in the rest of the cell.
All of the lysosomal enzymes are acid hydrolases. These digesting enzymes will only function properly in an environment with a pH of 5 (only inside a mature lysosome). Comparing to the neutral pH 7.2 in the cytosol, the pH 5 is two orders of magnitude more acidic. Even if the lysosomal membrane breaks down, the released acid hydrolases would be inactive at cytosol’s neutral pH. This design restricts the enzymatic activities only inside the lysosomes.
To maintain their acidic internal pH, lysosomes must actively concentrate H+ ions (protons). This is accomplished by a proton pump (called v-ATPase) embedded in the lysosomal membrane. The proton pump actively transports protons into the lysosome from the cytosol and requires an expenditure of energy in the form of ATP hydrolysis, since it maintains approximately a hundredfold higher H+ concentration inside the lysosome.
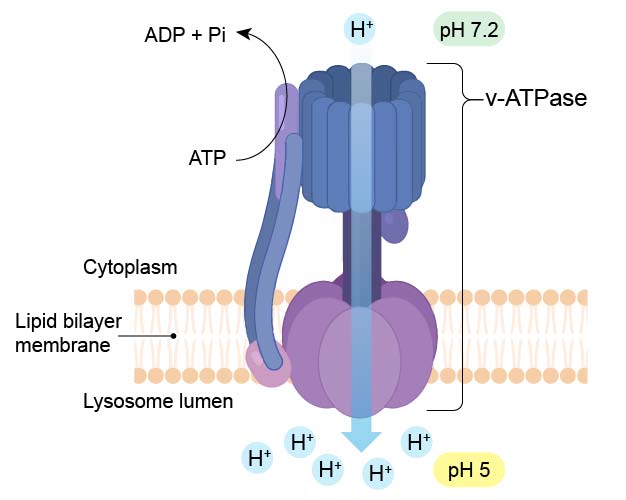
[In this figure] Diagram of v-ATPase which pumps H+ ions into lysosome to maintain a low pH (pH 5) environment.
Endocytosis and lysosome formation
Lysosome formation (or biogenesis) represents an intersection between the secretory pathway, through which lysosomal proteins are processed, and the endocytic pathway, in which extracellular molecules are taken up at the cell surface. Material from outside the cell is taken up by endocytosis and packed into endocytic vesicles, which bud from the plasma membrane and then fuse with early endosomes. The early endosomes gradually mature into late endosomes, which are the precursors to lysosomes. During endosome maturation, the internal pH is lowered to about 5.5, which plays a key role in delivering lysosomal acid hydrolases from the trans-Golgi network.
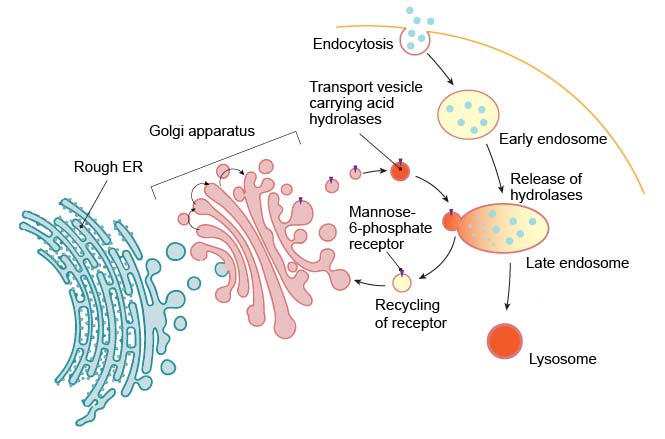
[In this figure] Lysosomes are formed by the fusion of transport vesicles budded from the trans-Golgi network with endosomes.
Enzymes of the lysosomes are synthesized in the rough endoplasmic reticulum (RER) and exported to the Golgi apparatus. The enzymes are trafficked from the Golgi apparatus in small vesicles. Enzymes destined for the lysosomes are specifically tagged with the molecules – “mannose 6-phosphate” (like a shipping label) so that they are properly sorted into acidified vesicles. These enzymes are recognized by mannose-6-phosphate receptors in the trans Golgi network. The acidified vesicles fuse with late endosomes, and the acidic internal pH causes the hydrolases to dissociate from the mannose-6-phosphate receptor. The hydrolases are thus released into the lumen of the endosome, while the receptors are eventually recycled to the Golgi. Late endosomes then mature into lysosomes as they acquire a full complement of acid hydrolases.
Lysosome function
Lysosomes act as the waste disposal system of the cell by digesting materials from both inside and outside the cell. Material from outside the cell is taken-up through endocytosis or phagocytosis, while material from the inside of the cell is digested through autophagy.
Cell eating and digestion – endocytosis and phagocytosis
Two ways for cells to take up materials from the outside: phagocytosis and endocytosis. Most cells perform endocytosis routinely. The molecules near or on the cell membrane’s surface could be internalized by the invagination of the cell membrane and formed a vesicle, called the endosome.
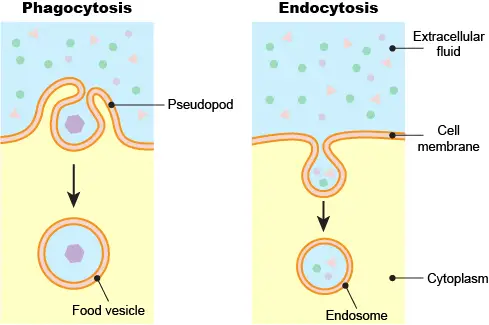
[In this figure] Phagocytosis vs. endocytosis.
Phagocytosis is common in single-celled eukaryotic cells to engulf larger food particles. Phagocytosis requires the cell to extend its pseudopods to surround the particle and bring it inside the cell as a food vesicle. Endocytosis does not involve the dramatic change of cell shape like phagocytosis. Instead, endocytosis initiates a local depression of the cell membrane to create an endosome.
Like we mentioned above, endosomes could be the precursors to lysosomes. Lysosomes are re-usable organelles. When new endosomes form, existing lysosomes can go and fuse with them to form endolysosomes. In endolysosomes, lysosomal enzymes degrade the contents from the endocytosis. After that, lysosomes could be re-used again.
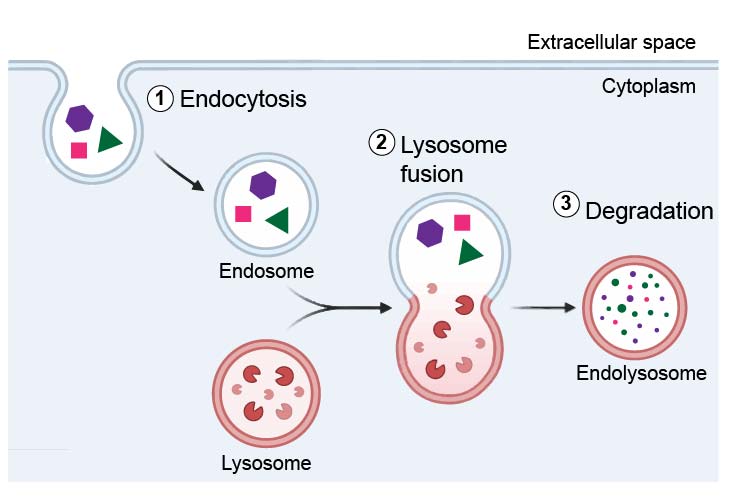
[In this figure] One of the major functions of lysosomes is to digest material taken up from outside by endocytosis.
Phagocytosis is carried out by specialized cells (e.g., macrophages) that engulf large extracellular particles, such as dead cells or foreign invaders (e.g., bacteria). The internalized particles fuse with the lysosome for degradation. Amino acids and nucleotides, products of lysosomal digestion, are recycled back to the cell for the synthesis of new cellular components.
Cell “self-eating” and autophagy
In the previous section, we mention lysosomes can recycle unwanted old cell parts. However, how does the cell decide what should be recycled in the lysosomes?
“Autophagy” (aka “self-eating”) is a biological process when cells are short of nutrient supply. When the cells sense that the environment becomes more challenging or the nutrient source is low, the cells turn on autophagy. To obtain the nutrient, the cells have to recycle some of their existing proteins and organelles, especially the misfunctioned ones.
The way our cells labeling the misfunctioned proteins is by putting “garbage tags” on bad proteins as an indication that “this guy should be recycled.” The “garbage tags” in cells is a small protein, called Ubiquitin. A group of enzymes performs the action of ubiquitination by adding Ubiquitin tags onto the bad proteins.
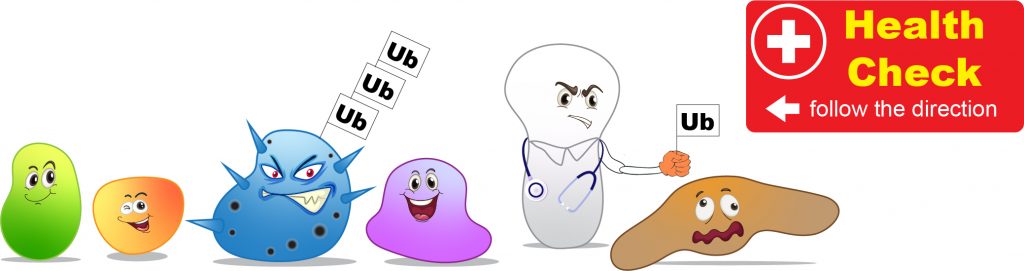
[In this figure] Cells screen for misfunctioned proteins constantly.
Once the bad proteins are founded, they will be labeled with a Ubiquitin (Ub) tag for degradation.
Cells go through their inventory at the beginning of the autophagy, sort out the bad proteins, and put the “garbage tags” on them. Nest, these enzymes in charge of autophagy will see these “garbage tags” and build the “garbage bags” to contain the cellular garbage. These “garbage bags” are the autophagosomes which consist of special lipid-formed vesicles.
The autophagosomes carry the cellular garbage to lysosomes and form an autolysosome, where the lysosomal enzymes break down the cellular garbage into raw materials. After that, cells can obtain nutrients or have ingredients to build new proteins and organelles.
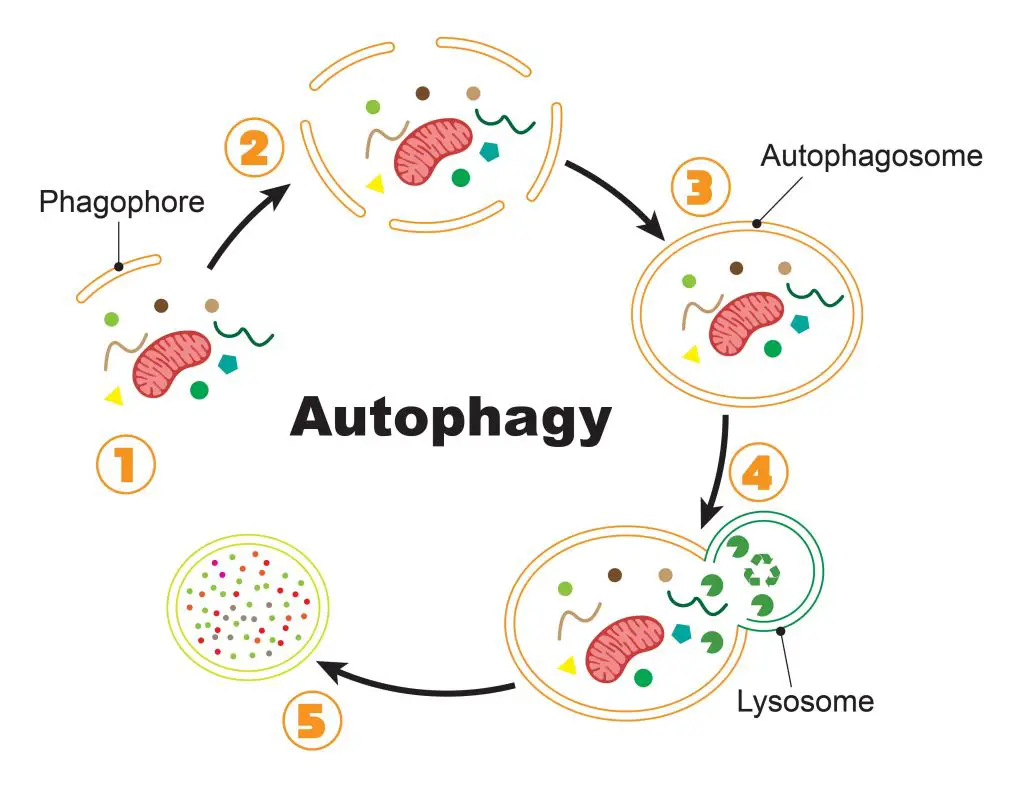
[In this figure] The process of autophagy.
(1) Once the cell initiates the autophagy, special lipid components (called phagophore) are recruited close to the bad proteins/organelles tagged for recycling. (2) More and more phagophores gather around the cellular garbage. (3) Phagophores assemble into a complete vesicle, called the autophagosome. (4) Fusion with the lysosome brings the digestive enzymes to break down the contents. (5) Bad proteins/organelles are recycled as raw materials for the cells to use.
Sometimes, even entire organelles can be packed into autophagosomes for recycling. For example, the special autophagy to degrade bad mitochondria is named “mitophagy.”
In 2016, the Nobel Prize in Physiology or Medicine was award to a Japanese cell biologist, Dr. Yoshinori Ohsumi. His discoveries opened the path of understanding the fundamental importance of autophagy in our cells. Now, we already know that many diseases, including cancer, Alzheimer’s, and Parkinson’s diseases, could be a consequence of misfunctioned autophagy.

[In this figure] The Nobel Prize in Physiology or Medicine 2016 was awarded to Yoshinori Ohsumi “for his discoveries of mechanisms for autophagy.”
Photo source: The Nobel Prize.
Lysosomes may do more than we know
Scientists admit that they don’t know everything about lysosome yet. Lysosomes may not just a place for cells to store digestive enzymes. In fact, scientists recently found lysosomes are the center in the cell to sense if the nutrient level is fine or too low. If lysosomes sense the food storage is in danger, they can send messages to nucleus to activate corresponding genes to regulate cellular metabolism.
The discovery of lysosome
Lysosomes (from the Greek: lysis, loosen and soma, body) were discovered by the Belgian cytologist Christian René de Duve in the 1950s. De Duve was awarded the 1974 Nobel Prize for Physiology or Medicine for his discovery of lysosomes and other organelles known as peroxisomes.

[In this figure] The Nobel Prize in Physiology or Medicine 1974 was awarded jointly to Albert Claude, Christian de Duve, and George E. Palade for their works in cell biology, especially for the discoveries of cell organelles, lysosome, peroxisome, and ribosome.
Photo source: The Nobel Prize.
Originally, De Duve had termed lysosomes the “suicide bags” or “suicide sacs” of the cells for their hypothesized role in apoptosis. However, it has since been concluded that they only play a minor role in cell death.
Not to be confused with lysosome – peroxisome, proteasome, vacuole, lysozyme, and liposome
There are a few biological terms that sometimes get confused with lysosomes. Let see what exactly they are:
Peroxisome is another membrane-bound cell organelle that may have similar sizes and spherical shape as lysosomes. However, peroxisome is more like a chemical laboratory in the cells and carries much specific redox (reduction-oxidation) reactions.
Proteasome is a protein complex that degrades unneeded or damaged proteins by proteolysis. We won’t call proteasome a cell organelle.
Vacuole is a membrane-bound organelle that functions as a storage space mainly in plant cells. Sometimes vacuole may contain digestive enzymes that carry out lysosomal functions.
Lysozyme is an enzyme found in bodily secretions such as tears, saliva, and milk. It functions as an antimicrobial agent by destroying bacterial cell walls. Lysozyme is nothing to do with lysosomes.
Liposome is a man-made spherical-shaped vesicle that is composed of lipid bilayer membranes. A liposome is heavily researched for drug and vaccine delivery.
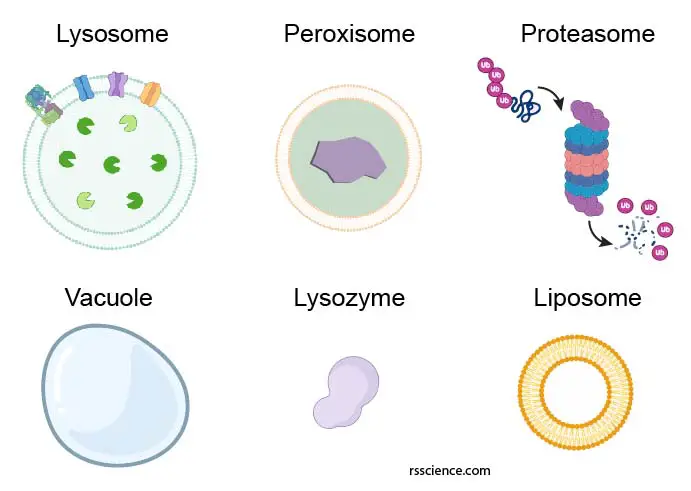
[In this figure] Representative diagram of the lysosome, peroxisome, proteasome, vacuole, lysozyme, and liposome.
Do plant cells have lysosomes?
Lysosomes were considered to be exclusive to animal cells. However, this statement became controversial. Discoveries in plant cells since the 1970s started to challenge this definition. Plant vacuoles are found to be much more diverse in structure and function than previously thought. Some vacuoles contain their own hydrolytic enzymes and perform the classic lysosomal activity, which is equivalent to autophagy. These vacuoles are therefore seen as fulfilling the role of the animal lysosome. However, the enzymes in plant’s vacuoles are different from animal’s lysosomes. Moreover, plant cells lack phagocytic functions. Therefore, it is not universally accepted that the vacuoles in plant cells are equal to lysosomes in animal cells.
Lysosome storage disorders
In humans, errors in the genetic code account for more than 50 lysosomal storage disorders. Most of these diseases result from deficiencies in single lysosomal enzymes. Due to the lack of certain enzymatic activity, undegraded material accumulates within the lysosomes of affected individuals and causes diseases.
The severity of a lysosomal disorder depends on which cells, tissues and organs accumulate the most toxic molecules. Many lysosomal disorders affect the heart, kidney, spleen, liver and/or bones. About half of these disorders also affect the central nervous system, which includes the brain and the spinal cord. Lysosomal disorders are chronic and progressive, meaning they worsen over time. People with these disorders may have life-limiting symptoms and a significantly shortened life span.
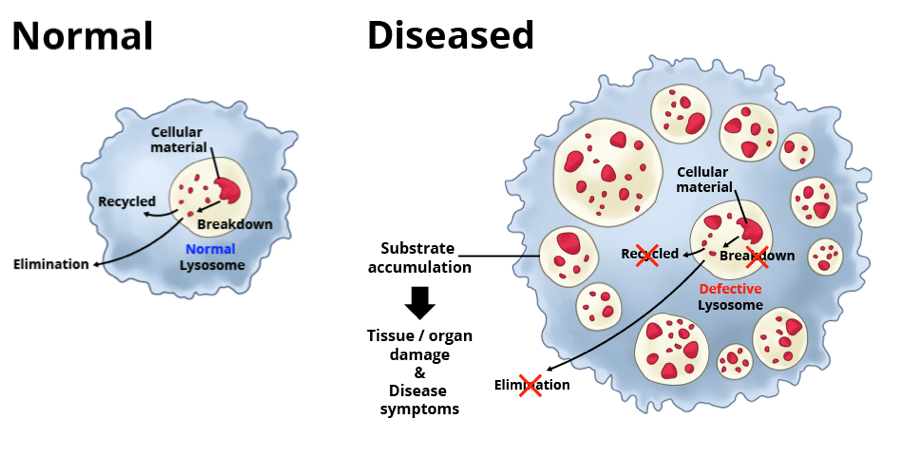
[In this figure] There are about 50 lysosomal disorders. In each, a different type of molecule builds up in the lysosome, disrupting normal cell function.
Photo source: Avrobio.
For example, Gaucher’s disease (the most common of lysosome disorders) results from a mutation in the gene that encodes a lysosomal enzyme required for the breakdown of glycolipids. The fatty substances accumulate in spleen and liver, causes these organs to enlarge and can affect their function. The fatty substances also can build up in bone tissue, weakening the bone and increasing the risk of fractures.
An intriguing exception is I-cell disease (inclusion cell disease), which is caused by a deficiency in the enzyme that catalyzes the first step in the tagging of lysosomal enzymes with mannose-6-phosphate in the Golgi apparatus. The result is a general failure of lysosomal enzymes to be incorporated into lysosomes. The secreted lysosomal enzymes are present in the blood of patients with I-cell disease, whereas their lysosomes are empty.
Summary
- Lysosome is a membrane-bounded sphere full of digestive enzymes and works as a recycling center in the cell.
- Lysosomal enzymes break down whatever substance entering the lysosomes into raw materials, such as amino acids, nucleotides, lipids, and sugars, so the cell can reuse these raw materials to build new organelles.
- Inside the lysosome is an acidic environment (pH 5), which activates the digestive enzymes. These enzymes can not be active in the cytosol (pH 7). This is a safety mechanism in the cell in case the lysosomes somehow leak or burst.
References
“Lysosomes” The Cell: A Molecular Approach. 2nd edition.
“Lysosome” British Society for Cell Biology
“Lysosomes as a therapeutic target”
“Signals from the lysosome: a control centre for cellular clearance and energy metabolism”